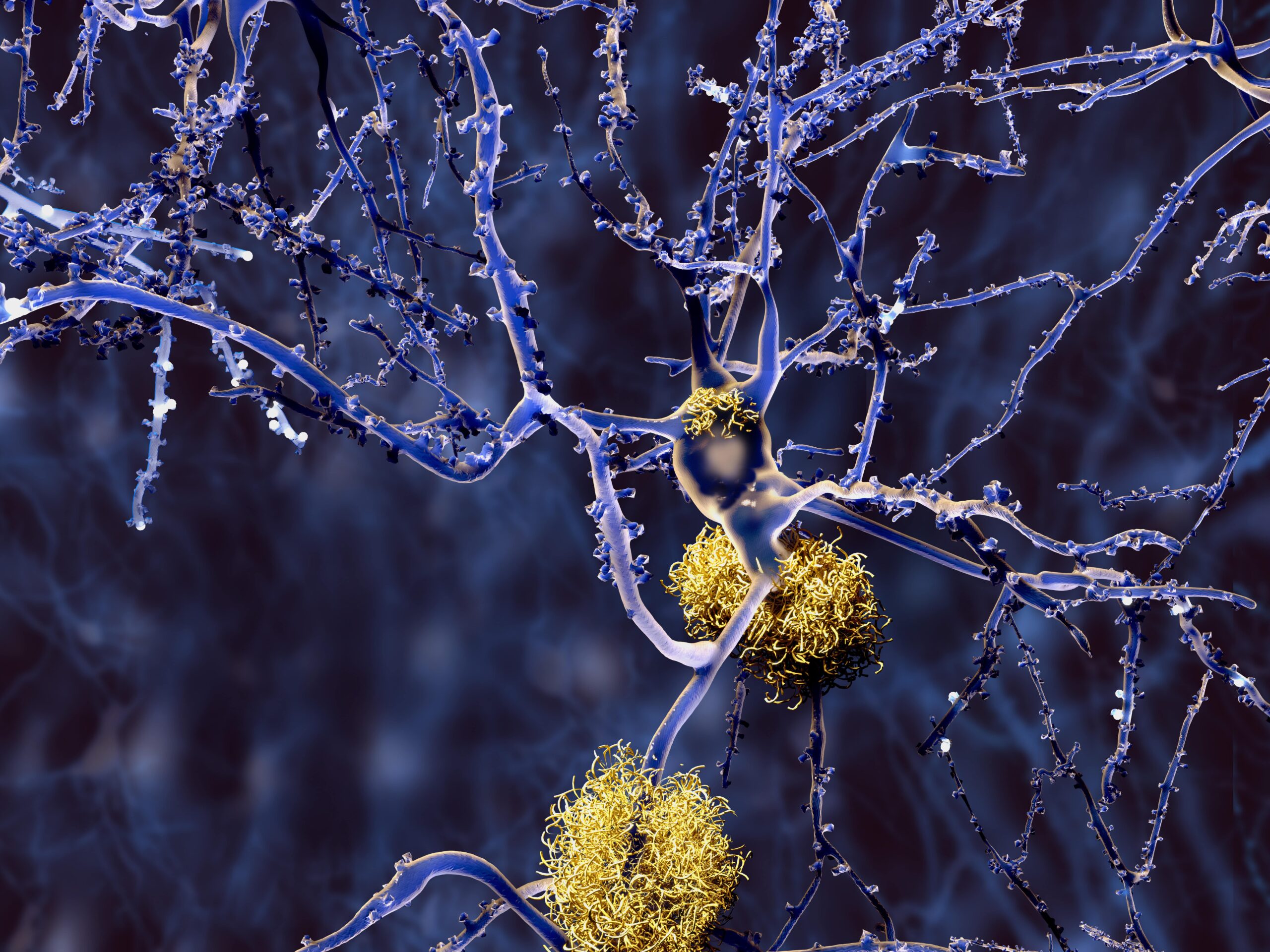
Neurodegenerative diseases such as Alzheimer’s disease are known to be linked to the accumulation of amyloid-β plaques, ultimately disrupting cognitive function. Now, a study conducted by researchers from Weill Cornell Medicine has identified a surprising culprit that could contribute to the buildup of these toxic proteins: CD36 receptors on the surface of macrophage cells in the brain. These findings add critical information to our understanding of how neurodegeneration develops and progresses in conditions like Alzheimer’s disease (AD), Parkinson’s disease, and cerebral amyloid angiopathy (CAA), and, importantly, may lead to better treatment options and outcomes for patients in the future.
The Link Between Neurodegeneration and Amyloid Plaques
While the exact cause of Alzheimer’s disease is unknown, for years, scientists have observed that abnormal amyloid-β accumulation is one of the defining characteristics and hallmark early indicators of AD.
In healthy brains, amyloid-β is a regular byproduct of cognitive functioning and is cleared away by immune cells. But in AD and CAA, this process is impaired: amyloid-β aggregates and forms plaques. These plaques disrupt the brain’s neural network and lead to irreversible neurodegeneration. However, despite this known association, researchers have long struggled to understand what triggers this buildup. In recent years, some researchers have turned their focus toward uncovering this trigger. And in a groundbreaking new study, a team of researchers from Weill Cornell Medicine may have identified it.
CD36 Receptors Implicated
Previous research has demonstrated that border-associated macrophages, immune cells surrounding blood vessels in the brain, have CD36 receptors on their surface. Research has also previously shown that amyloid-β can bind to these receptors.
The Weill Cornell Medicine team’s study, published in Molecular Degeneration, found that, in fact, amyloid-β does not just bind to CD36 receptors — it also activates them. The team found that, when activated, these CD36 receptors released a flood of toxic free radicals, paralyzing brain blood vessels.
Next, as study senior author Dr. Costantino Iadecola, director and chair of the Feil Family Brain and Mind Research Institute and the Anne Parrish Titzell Professor of Neurology at Weill Cornell Medicine, explains, “Amyloid then accumulates in the smooth muscle of the vessel walls, which blocks the vessel’s ability to pump blood to the brain. CAA and cognitive impairment are the unfortunate result.”
The team arrived at this discovery by using preclinical models of AD with CAA and cognitive impairment. They eliminated CD36 receptors from these Tg2576 mice, and found that doing so led to several critical outcomes: it suppressed oxidative stress, improved neurovascular functioning, decreased amyloid-β accumulation, and improved overall cognitive functioning.
Ultimately, the team’s findings may highlight a critical pathway in the pathogenesis of AD. Amyloid-β triggers CD36 receptors to produce free radicals. Free radicals harm arteries, leading to abnormal amyloid-β deposits on arterial walls. These deposits injure arteries, suppress critical blood flow to the brain, and ultimately worsen cognitive decline.
Further research will help determine if blocking or manipulating CD36 macrophage receptors could eliminate abnormal accumulations of amyloid-β in patients with diseases like AD, CAA, and other conditions associated with amyloid accumulation in blood vessels. The team hopes that this future research may lead to finding better treatment options for patients with AD.
Iadecola and his team believe that these findings could be significant for patients with amyloid-related imaging abnormalities (ARIA), a potentially severe reaction to certain monoclonal antibody drugs — such as aducanumab — used to treat patients with AD and mild cognitive impairment. At present, patients with ARIA must discontinue treatment with these drugs because of the risk of brain edema or brain hemorrhages. Therapies targeting CD36 macrophages may lead directly to more treatment options for patients with ARIA and may result in more successful amyloid-β immunotherapies for patients with related conditions.
Scantox is a part of Scantox, a GLP/GCP-compliant contract research organization (CRO) delivering the highest grade of Discovery, Regulatory Toxicology and CMC/Analytical services since 1977. Scantox focuses on preclinical studies related to central nervous system (CNS) diseases, rare diseases, and mental disorders. With highly predictive disease models available on site and unparalleled preclinical experience, Scantox can handle most CNS drug development needs for biopharmaceutical companies of all sizes. For more information about Scantox, visit www.scantox.com.